前沿进展:通过层次结构信息和分子动力学模拟发现高活性肽
应用介绍
设计一种新多肽药物,就像在化学和生物的迷宫中找到一条最有效的路径。多肽药物在治疗中具有广泛的应用潜力,但如何从众多可能的多肽序列中快速找到高效的活性肽?目前的方法如机器学习等,往往面临高昂的计算资源消耗、复杂的亲和力评估以及数据要求极高的局限。为了解决这些问题,基于算法信息论框架下的“梯径(Ladderpath)”方法,提出了一种创新的多肽设计策略——PepHiRe (Peptide Hierarchical Reconstructor)。这一方法可以将多肽序列的层级结构信息进行解析,通过快速高效的方式生成潜在的高活性肽药物。借助PepHiRe,我们能够更清晰地理解多肽药物的结构复杂性,加速从设计到实际应用的过程。
多肽药物是由2到50个氨基酸通过肽键连接而成的小分子。相比于传统的小分子药物,多肽药物具有更高的特异性和更低的毒性,这使得它们在治疗癌症、糖尿病以及心血管疾病等多种疾病方面展现出巨大的潜力。每一个多肽序列都有其独特的信息,就像在药物设计的“地图”上占据着特定的位置。这些位置代表着多肽的特性,例如它的靶向性、药效和稳定性。
然而,所有可能的多肽序列种类是几乎无穷的。仅考虑由20种天然氨基酸组成的多肽序列,可能的组合数量就远远超出想象。这意味着,探索所有潜在多肽药物的可能性就像试图浏览一张无比巨大的地图。我们很难完全覆盖整个多肽药物的设计空间,所以需要通过系统性的方法,帮助高效地探索局部区域,从中找出对疾病治疗具有实际意义的多肽分子。
为了解决这些问题,需要探索更加智能化的多肽设计方法。我们基于“算法信息论”中的“梯径(Ladderpath)”方法提出的PepHiRe方法,是一种通过层级结构解析,帮助研究者在复杂多肽序列空间中找到最优多肽药物的创新工具。
目前多肽药物设计的方法 在多肽药物设计中,有几种常见的计算方法,分子对接用于预测肽与靶蛋白的相互作用,类似于为钥匙寻找合适的锁,从而筛选出可能有效的肽药物。分子动力学模拟(MD)则帮助研究人员更深入地观察多肽在生物环境中的动态行为,比如与靶蛋白的结合过程和结构变化。虽然这些方法有效,但在使用时往往耗费大量的计算资源,且在处理复杂分子时效率不高。 近年来深度学习也进入了多肽药物设计领域。通过神经网络,研究者可以自动生成新肽分子,像是让机器学习如何“设计”药物。然而,深度学习方法常常需要大量高质量的数据来训练模型,而且由于其“黑箱”特性,研究人员很难理解模型的内部工作原理。
在自然界中,复杂事物的进化通常会依赖于已有的基础进行“修修补补”,而不是从零开始。复杂的器官、分子或功能大多是在已有结构上不断优化与调整 [1] 。基于这一观察,我们提出了PepHiRe,结合“算法信息论”中的梯径(Ladderpath)”方法,系统性地设计和优化多肽药物。PepHiRe就像是在已有的多肽序列中寻找关键的“拼图”,然后通过重组这些有用的片段,生成新的多肽药物,再通过一些虚拟筛选的工具帮助我们验证生成的多肽序列的有效性。
梯径方法基于算法信息论的核心理念:任何复杂系统都可以通过找到其中重复的部分进行简化和优化。就像自然进化不是从头开始,而是通过重复和调整已有的片段来构建新的结构,多肽序列也是由一些关键片段反复组合而成的 [2] 。通过梯径方法,可以识别这些关键的片段及其层次关系。就像搭建积木一样,使得可以不从零开始构造多肽,而是基于已知的有效片段进行组合和优化,生成更加优化的多肽药物。
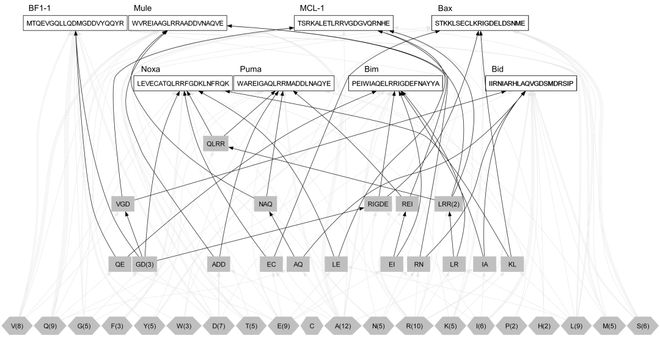
PepHiRe利用这一思想,从8种已知的BH3肽序列中提取出结构信息,辅助生成全新的多肽药物,进而靶向抑制与癌症密切相关的MCL-1蛋白。MCL-1蛋白通过阻止细胞凋亡,帮助癌细胞“逃脱”死亡,而我们设计的多肽药物则可以模仿BH3肽的功能,打破这种抑制,从而促进癌细胞死亡,减缓病痛。
① 生成新肽序列:利用梯径方法从8种已知的高效BH3肽序列中提取结构信息,生成一系列新的多肽候选序列。这些新生成的多肽序列都具有潜在的与MCL-1结合能力。
② 筛选多肽:通过预测这些多肽的螺旋结构潜力(即它们能否形成α螺旋),与目标蛋白分子对接结果来筛选出表现最佳的多肽。通过这一过程,我们能够选出与MCL-1结合最紧密的多肽。
③ 优化与迭代:筛选出的最优多肽会被加入到原始的多肽库中,形成一个扩展版的多肽库,供下一轮生成和筛选使用。这个过程会不断重复,以确保最终生成的多肽具备高度的生物活性和靶向效果。
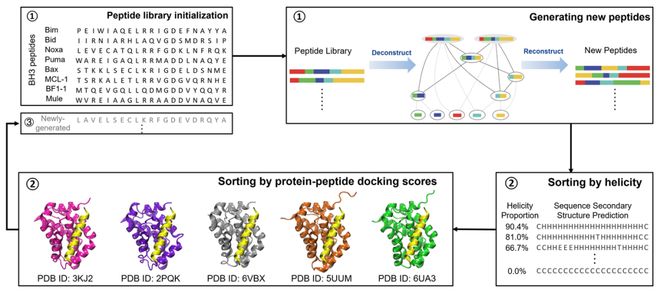
我们可以将梯径方法想象成一个“拼图”过程,它从已知的多肽序列中提取出有用的片段,称为“梯元(ladderon)”,并将它们重新组合成新的多肽序列。
首先,生成一个“梯图(laddergraph)”,展示哪些片段在原始序列中反复出现。每个片段的权重由其长度和出现频率决定,权重越大,越有可能被优先选中。然后,我们初始化一个空的多肽序列,使用轮盘赌的方式,根据这些片段的权重,逐一将它们放入合适的位置。
一个关键点是,如果选中一个较大的片段,会将其放置在仍有空位的位置,该片段会完全覆盖这些位置中的片段 (如下图中ABDCD覆盖了DCD) ;但如果指定的位置已经完全被占用,则跳过该片段。这一操作确保了序列中较高层次的梯元 (ladderon) 具有更重要的意义。通过这种“拼图”过程,我们不仅能够高效生成新肽序列,还能够确保关键的功能片段被保留下来。
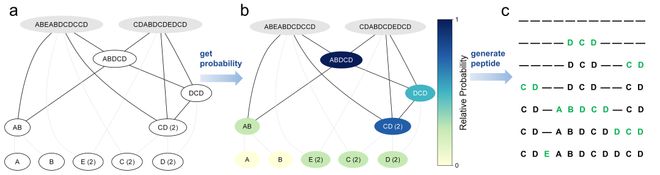
生成了大量多肽序列后,需要找到最有潜力的多肽来继续优化。首先,利用PsiPred工具预测这些多肽的螺旋结构,因为在与MCL-1的结合中螺旋性起着重要作用。随后,挑选出螺旋性最好的肽,并通过MODPEP生成它们的三维结构,再使用HDock进行蛋白对接分析。HDock能够快速高效评估多肽与MCL-1的结合效果。
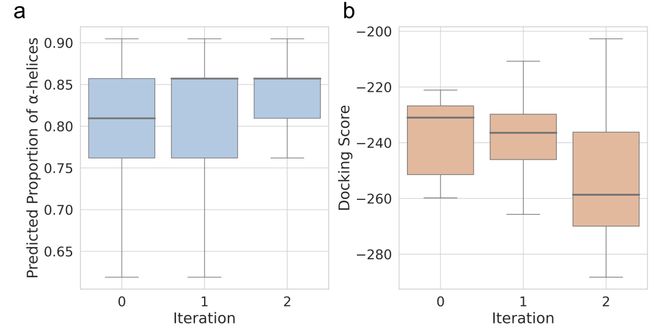
当选出表现最好的多肽,会将其加入原始多肽库,形成一个扩展版的多肽库,用于下一轮生成和筛选。随着每次迭代,多肽的螺旋结构逐渐增强,对MCL-1的结合能力也在逐渐提升如下图。最终,经过几轮优化,我们成功筛选出了与MCL-1结合更紧密、螺旋性更强的多肽序列。
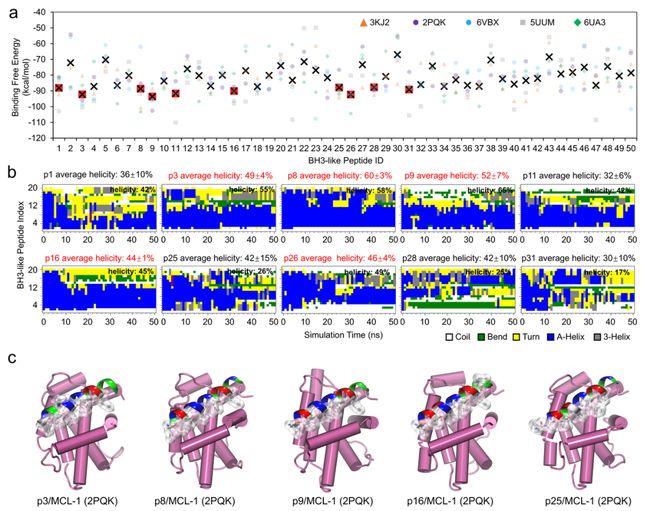
为了更好地了解生成多肽与MCL-1的结合的稳定性,我们从PepHiRe生成的多肽中挑选了50条表现最好的多肽进行分子动力学 (MD) 模拟。每条多肽会分别与5个不同结构的MCL-1蛋白对接,并进行了50ns的模拟,以确保它们的结合在不同条件下都足够稳定。
结果表明,大多数多肽的结合结构在模拟过程中达到了稳定状态。随后,我们使用MM/GBSA方法计算了这些肽的结合自由能,前10名多肽的结合力表现尤为突出,说明它们与MCL-1的结合非常紧密。
之后对结合最好的10条肽进行了进一步的结构模拟,发现这些多肽在水中依然保持较高的螺旋性。这种螺旋结构与MCL-1的结合槽非常契合,进一步说明了它们的抑制作用。有趣的是,这些多肽展现了典型的“疏水-亲水”双面结构,很好的模拟了天然BH3蛋白与MCL-1的结合方式,证实了它们在调控MCL-1凋亡通路中的潜力。
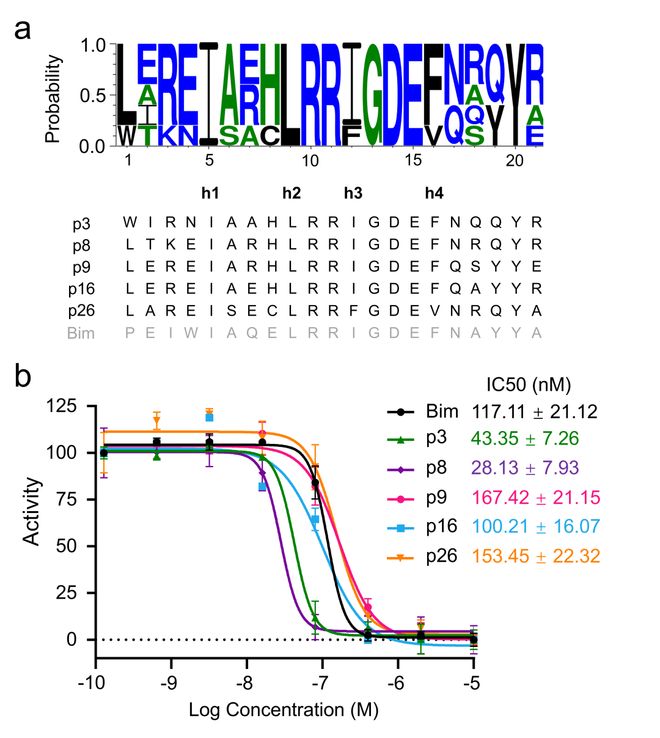
为了验证筛选出的BH3样肽的实际效果,我们合成了五条在MD模拟中表现最好的肽 (p3、p8、p9、p16和p26) ,并进行了真实的生物实验。通过荧光偏振 (FP) 实验,这些肽展示了强效的MCL-1抑制作用,IC50值(即半数抑制浓度) 在28.13到167.42 nM之间。特别是其中的三条多肽 (p3、p8、p16) 的抑制效果甚至优于天然的Bim BH3肽 (117.11 nM) 。这些实验结果表明,PepHiRe设计的多肽不仅在模拟中表现优异,在真实生物实验中也展示出了强大的抑制潜力,证明了它们作为MCL-1抑制剂的有效性。
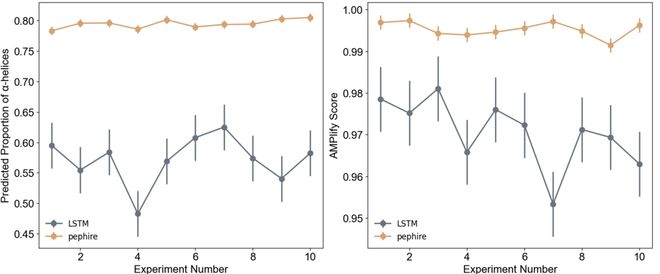
为了展示PepHiRe的广泛应用,我们还将其用于设计Magainin抗菌肽。Magainin是一种能够破坏细胞膜的抗菌肽。我们从数据库中收集了95条相关序列,并通过PepHiRe生成了1万条新肽。使用PsiPred工具筛选出螺旋性超过90%的肽后,利用AMPlify模型进一步评估它们的抗菌效果,最终生成了500条具有增强抗菌活性的肽。
与传统的深度学习方法 (如LSTM RNN) 相比,PepHiRe不仅生成的多肽在螺旋性和抗菌活性上表现得更好 (如图) ,而且计算资源需求更少,速度更快。PepHiRe只需一个CPU,而LSTM RNN则需要更强的GPU支持。
PepHiRe与经典方法的对比 在广阔的多肽空间中寻找有效序列是一项复杂的任务,PepHiRe通过缩小搜索范围,提高了效率。为验证其效果,我们也将PepHiRe与两种经典方法进行了比较:单点突变法和遗传算法(GA)。 单点突变法通过对已有多肽序列的单个氨基酸进行随机突变生成新多肽。对于同样的任务,从8条已知BH3多肽序列开始,经过迭代生成新序列,同PepHiRe一样也会选出得分最高的多肽加入初始库。结果显示虽然评分有所提升,但新多肽的螺旋结构并未显著改善,且生成的多肽与原始多肽非常相似,仅在少数位置发生变化,说明此方法的探索范围有限,难以有效扩展到更广泛的多肽空间。 遗传算法通过模拟生物进化生成新多肽,具体操作包括交叉和突变,生成并筛选新序列。从8条已知BH3多肽序列开始,虽然螺旋性在迭代过程中逐步提高,但对接评分的提升并不显著,表现完全不如PepHiRe或单点突变法。虽然生成的多肽序列可能比单点突变法更具多样性,但与PepHiRe相比仍显不足。另外,遗传算法需要对更多的多肽进行对接评分,运行速度比其他方法慢了数倍。
PepHiRe是一种基于梯径(Ladderpath)方法的创新工具,能够从已有多肽序列中提炼关键信息,帮助我们设计出功能强大的新型多肽药物。它通过模拟自然进化的过程,从现有多肽中提取出最重要的部分,重新组合为具有生物活性的全新多肽。与依赖大量数据的机器学习方法不同,PepHiRe即使在数据有限的情况下也能高效工作,特别适合早期药物设计中的复杂任务。
未来,PepHiRe不仅可以用于MCL-1抑制剂的设计,还可能扩展到其他多肽和蛋白质领域。通过不断优化和扩展,我们期待这种方法能够在药物设计、结构预测等领域发挥更广泛的作用。
关于代码和数据 PepHiRe的源代码可在免费获取。其中还包括MD模拟的输入文件和输出轨迹、初始配置、拓扑结构、参数、模拟条件以及相应的轨迹文件。
大模型、多模态、多智能体层出不穷,各种各样的神经网络变体在AI大舞台各显身手。复杂系统领域对于涌现、层级、鲁棒性、非线性、演化等问题的探索也在持续推进。而优秀的AI系统、创新性的神经网络,往往在一定程度上具备优秀复杂系统的特征。因此,发展中的复杂系统理论方法如何指导未来AI的设计,正在成为备受关注的问题。
集智俱乐部联合加利福尼亚大学圣迭戈分校助理教授尤亦庄、北京师范大学副教授刘宇、北京师范大学系统科学学院在读博士张章、牟牧云和在读硕士杨明哲、清华大学在读博士田洋共同发起,探究如何度量复杂系统的“好坏”?如何理解复杂系统的机制?这些理解是否可以启发我们设计更好的AI模型?在本质上帮助我们设计更好的AI系统。读书会于6月10日开始,每周一晚上20:00-22:00举办。欢迎从事相关领域研究、对AI+Complexity感兴趣的朋友们报名读书会交流!